Annals of Neurosciences, Vol 15, No 3 (2008)
Annals of Neurosciences, Volume 15, Issue 3 (July), 2008
FUNCTIONAL MODULATION OF THE P53 GENE AND ITS PROTEIN IN HUMAN BRAIN TUMORS
Corresponding author:
Dr. Rajalakshmi Gope
Additional Professor
Department of Human Genetics
National Institute of Mental Health and Neurosciences (NIMHANS)
Bangalore 560 029.
Phone: 091802699 5125
Fax: 091 80 2656 4830
Email:
Alternate email:
(Date received: 4/4/2008)
Abstract
The primary human brain tumors account for less than 2% of all human cancers but yet cause a disproportionate burden of cancer related morbidity and mortality. These are the second most common form of tumors in the pediatric population, next only to leukemia and hence the second leading cause of death due to cancer in children. Among the adults they rank 6th and 8th in frequency of all neoplasms and form 2nd and 5th leading cause of death in men and women respectively who belong to the age group of 20 to 39 years. The survival rates for brain tumor patients have not changed over the past several decades. The p53 gene is one of the most important and intensively studied human tumor suppressor genes. It has been shown to play a major role in cell proliferation as well as cell death. Because of its varied functions the p53 gene and its pathways have become important therapeutic target. Mutations in the p53 gene and the oncogenic function of its protein product have been well documented. However, numerous evidences indicate that based on its conformation and post translational modification the function of the wild type p53 protein can be modulated to be similar to that of the mutant form. In this paper we have reviewed the functional modulation of p53 gene in human brain tumor development.
Keywords : Brain tumors, Cell-cycle, Conformation, p53 protein, Ser-392 phosphorylation, Tumor suppressor gene
Introduction
Human brain tumors arise from various cells of the central nervous system and the nomenclature is based on the origin of cell type (Figure 1). The tumors that arise from the glial cells are called gliomas. However, most gliomas are a heterogeneous group of tumors that arise from various cell types (1). Tumors that arise from the cells resembling the primitive neuroepithelium, that is, the precursors of the nervous system are called embryonal or primitive neuroectodermal tumors (PNETs). Medulloblastomas are the most common among the PNETs (1,2). The most prevalent primary brain tumors among the pediatric population are astrocytomas, ependymomas and medulloblastomas. Some of the common adult brain tumors include diffuse astrocytic tumors such as astrocytomas, anaplastic astrocytomas and glioblastoma multiforme (GBM). The oligodendrogliomas and meningiomas are also some of the commonly found brain tumors in adults (3).
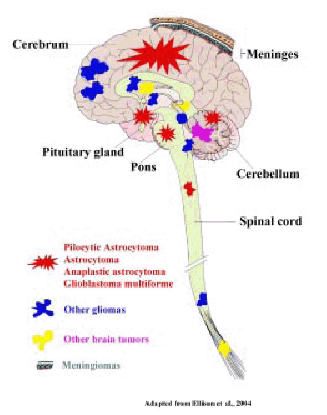
Figure 1. Diagramatic representation of the location of vurious human brain tumor types.
Types of Human Brain Tumors
Gliomas
Gliomas are the most common form of brain tumors accounting for more than 50% of all primary brain tumors and more than 90% of all primary malignant central nervous system (CNS) tumors (4,5). Astrocytomas account for approximately 80% of all malignant brain tumors and are the most prevalent among the glial tumors. Approximately 25% of gliomas are oligodendrogliomas or mixed oligoastrocytomas. Gliomas are prevalent in both pediatric as well as in adult population (1, 2). In general gliomas affect males 40% more frequently than females. Male predominance is evident in adolescent population and this observed male predominance does not exist if the tumor frequency is compared between the older males and the females who have crossed the menopause. This data is suggestive of a protective role for female hormones against this tumor type (6).
Meningiomas
Meningiomas are the second most common form of human brain tumors. These are also the most common non-glial primary intracranial tumors constituting approximately 20% of all intracranial neoplasms. Most meningiomas occur in adults of middle and old ages. These tumors are uncommon in pediatric population. Meningiomas in a benign form show a female preponderance, with a female to male ratio of approximately 2:1. However, among the malignant meningiomas the female to male ratio is 1:1 (1,2).
Other Brain Tumors
The medulloblastomas are the second most common pediatric brain tumors and also the most common malignant brain tumors found in children. The age of peak incidence is between 5 and 9 years and a second peak occurs in adults from 20 to 24 years (1,2).
Hemangioblastomas are tumors of uncertain cell origin comprising approximately 2% of primary central nervous system tumors and 25% of these tumors occur in association with the von Hippel-Landau (VHL) syndrome. These are adult tumors that primarily occur between the ages of 30 and 60 years (7). Gangliogliomas are benign tumors composed of a mixture of neoplastic astrocytes and nerve cells and they predominantly affect infants or young children of both sexes equally. Craniopharyngiomas arise from the embryologie precursors of the anterior pituitary gland and occur from infancy to old age, with the predominant peak in childhood at 5 to 14 years and in adults at 65 to 74 years (1,2).
Symptoms
Most brain tumors present in one of the following ways: seizures, focal signs and signs of increased intracranial pressure (ICP). ICP is commonly presented in any of the following forms such as headache, typically frontal or generalised and worse on waking, nausea and vomiting and reduced conscious level. The factors that determine the signs and symptoms of brain tumors generally include the location, growth rate, histology and size of the tumor (7). Presence of papilledema and optic atrophy is suggestive of raised intracranial pressure. Early symptoms are often non-specific such as forgetfulness, word-finding difficulties, headaches and in many cases a history going back several months is obtained in retrospect. By the time of presentation, however, clinical signs are usually obvious and even basic examination of the motor or sensory systems, speech function and visual fields will reveal significant abnormalities (4).
Etiology
Certain inherited genes or exposure to certain physical or chemical agents are considered to be potent risk factors which could play a causal role for brain tumor development (2). Some of the genetic polymorphisms that are specific for detoxification or metabolic enzymes are implicated in the pathogenesis of certain brain tumor types. Data from some studies have suggested mutagen sensitivity to gamma radiation and exposure to therapeutics, diagnostic or industrial ionizing radiation to be significantly associated with risk of brain tumor development (8). A personal medical history of serious head trauma and also seizures have been implicated as potential risk factors Various factors like diet containing cured food items, alcohol, tobacco, residential chemicals like N-nitroso compounds, industrial and occupational exposure to various carcinogenic and neurotoxic substances are also suspected to act as causal agents for brain tumor development (1,2).
Classification and Grading
In 1979, the World Health Organization (WHO) published a classification system that encompassed all the central nervous system tumors and they were classified based on the cell or tissue of origin and their patterns of differentiation. It was subsequently revised in 1993, which included tumor data from immunohistochemistry (9). The WHO classification system includes 7 major categories and more than 120 sub categories of brain tumors (3,10,11). They include tumors of the neuroepithelial tissue, meninges, peripheral nerves, lymphomas and hemopoeitic neoplasms, germ cell tumors, sellar region and the metastatic tumors, to mention a few.
Histopathology
The histopathology of brain tumors varies widely between and within the tumor types (Figure 2). The pilocytic astrocytomas have diverse morphological spectrum which makes the histopathological diagnosis extremely difficult (2,3,9,11). The grade II astrocytomas show increased cellularity and microcytic formation. The grade III anaplastic astrocytomas exhibit increasing degree of anaplasia exhibiting complex nuclear morphology with increasing cellularity. The histopathology of the GBMs is highly variable. They show a high degree of cellular and nuclear pleomorphism and numerous multinucleated giant cells (9). The classic Oligodendroglia tumor is moderately cellular composedof cells with roundnucleiand perinuclear halos (9,11). The typical histologic features of grade I benign meningiomas include a whirling pattern, concentric calcifications known as psammoma bodies,and nuclear pseudo-inclusions(9).
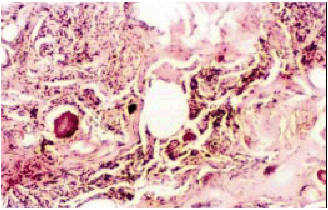
Figure 2 a : H & E stained section of transitional meningioma grade III.
Fibroblatic appearance, psammoma bodies and whorls of cells are seen. Magnification-100 X
Figure 2b : H & E stained section of pituitary adenoma. Magnification-100X
Genetic Alterations in Brain Tumors
A variety of chromosomal alterations have been reported for the various human brain tumor types(12). Allelic loss on the q and p arms of chromosome 17 including the p53 and NF1 gene loci have been shown in pilocytic astrocytomas grade I (13). Gain of chromosome 7 and 8 have also been reported in these tumors (14). Loss of heterozygosity (LOH) on chromosomes 6q, 13q and 22q has been reported in some astrocytomas. More than 60% of grade II astrocytomas have loss of alleles on 17p, including the p53 gene locus, and in these cases the remaining p53 allele was found to be mutated (15). LOH of the intron I regions of RB1 and p53 genes have been reported in brain and nervous system tumors (16–22). Amplification and over expression of PDGF and its receptor have been reported in all grades of gliomas and is considered to be an early event in glioma development (12,23). In glioblastomas mutations of the p53 tumor suppressor gene and EGF-R amplification/over expression are mutually exclusive events (24). A tumor suppressor gene frequently inactivated in astrocytic tumors is the CDKN2, which encodes the protein pl6 (25). pl6 protein binds to and inhibits the function of cyclin dependent kinase 4 (CDK4). CDK4 forms complex with cyclin D and inhibits pRb, the protein product of the tumor suppressor gene retinoblastoma (RB1) and results in loss of RB1-mediated growth suppression. Inactivation of either pl6 or RB1 is observed in most GBMs and rarely both are inactivated in the same tumor (26). RBI mutations have been reported in 25% of high-grade astrocytomas (27). LOH at the intron I region of the RB1 gene have been reported in brain tumors (19,20,21). Deletion or loss of an entire copy of the chromosome 10 is also a common finding in high-grade astrocytic tumors. Mutation as well as LOH of PTEN, a tumor suppressor gene located on chromosome 10q has been reported both in anaplastic astrocytoma and GBMs. Cytogenetic analysis has revealed abnormalities in the chromosomal copy number in chromosomes 1, 6, 7, 9, 10,13, 17,19 and 22 in many human brain tumor types (3). The most common chromosomal abnormalities associated with the meningiomas are at chromosome 22q locus. Chromosomal gains, most commonly on 20q, 12q, 15q, 1q, 9q and 17q, have been noted in high-grade meningiomas. In addition, alteration at the p53 tumor suppressor gene has been considered as a reliable marker for malignant transformation of meningiomas (2,3,21).
Diagnosis and Management
Computerized tomography (CT) and magnetic resonance imaging (MRI ) are the key diagnostic modalities for identification of human brain tumors. Positron emission tomography (PET) scanning and single photon emission tomography (SPECT) have supplementary roles in the imaging of brain tumors (28). A CT head scan with contrast will usually make the diagnosis of a brain tumor with some degree of certainty. Standard T1-and T2-weighted MRIs detect brain tumors with high sensitivity (7). There is some evidence that advanced MR techniques such as spectroscopy, to look at the chemical constituents of the tumor tissue, are used to improve accuracy in noninvasive diagnosis of tumor type and grade (29). For the present, however, the key to further management of almost all patients with brain tumors remains the establishment of an exact histological diagnosis. While an operative procedure is being arranged, majority of the patients are commenced on dexamethasone either orally or intravenous for management of tumor-related ICR Dexamethasone can very significantly reduce focal neurological deficit (30). The primary modality of treatment for most primary brain tumors is surgery. The key aim of surgery with many brain tumors is the achievement of a histological diagnosis (28). Radiation is used when the entire primary tumor cannot be surgically removed. Local radiation therapy techniques, including external focal, brachytherapy, and stereotactic radiosurgery, may be administered to patients (7). Highly focused radiotherapy has a role in the treatment of certain well-localised tumors; for example, stereotactic radiosurgery (with a Cobalt-60 “gamma knife”) is very effective with small targets (tumor size of below 3 cm diameter) and is also used in the management of tumor metastases (31). An alternative approach to very focal radiotherapy is interstitial brachytherapy with 125-iodine (2,4,7). Patients with certain tumor types show good response to drugs like Procarbazine, CCNU and Vincristine, Temozolamide and Liposomal Daunorubicin etc (2,4,7). Alternative approaches to delivering the drug to the tumor include intra-arterial chemotherapy and the implantation of wafers impregnated with Carmustine (BCNU), one of the first and still one of the most effective anti-glioma agents, directly into the resection cavity at the time of surgery (2,4,7).
Therapeutic attempts using the wild type p53 gene or restoration of its tumor suppressor function are underway (32). Various antiangiogenic drugs are under development; analogues of thalidomide inhibit the angiogenic action of bFGF and these may be seen in clinical trials over the next decade (33). The potentially interesting technique of Boron Neutron Capture Therapy is undergoing evaluation for treatment of malignant gliomas in a number of centres at present. Research in this area has lead to the use of nanotechnology (34,35). Targeted multifunctional polymer nano particles that specifically destroy brain tumors in laboratory animals have already been developed and their potential as a therapeutic in human needs to be evaluated (36).
The p53 gene
The p53 gene product was originally considered as an oncogenic protein. One of the evidences to disprove p53 as an oncogene came from the Friend virus induced mouse erythroeukaemias where p53 gene was found to be a frequent target for viral integration, which resulted in its inactivation (37). However, the most important finding came from the examination of the p53 clones used in the earlier transfection studies which revealed that the p53 gene used in these studies was actually a mutant form (38,39). Subsequent studies showed that p53 was indeed a tumor suppressor gene. By the year 1990, p53 was widely accepted as a tumor suppressor gene and was recognized as one of the frequently inactivated gene in more than 50% of all human cancers (40,41) (Figure 3).
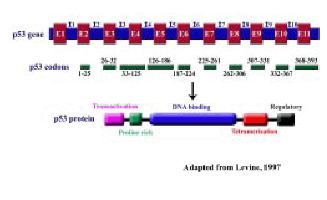
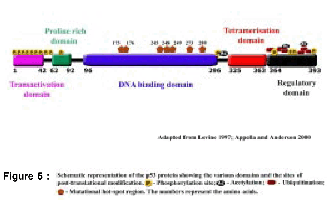
Figure 3 : Structure of the p53 gene and protein (Drawn not to scale): I-Intron; E-Exon
Structure of the p53 Gene and mRNA
The human p53 gene is located on chromosome 17pl3.1 (42). Southern blot analysis revealed presence of a single p53 gene in the human genome (43). The gene spans 20 kilobases (kb) of genomic DNA, and the structure is conserved in all the species studied. It comprises 11 exons, first exon is a non-coding exon, followed by a large first intron 10 kb in length (45) (Figure 3). There are five highly conserved domains within the exons 5, 7 and 8. The p53 mRNA transcript is approximately 2.8 kb in length (46) (Figure 3) and can be detected in most human cells, with the exception of cells in GO phase (47).
RFLP analysis of Genomic DNAs from several human tumor tissues with Bgl II restriction enzyme digestion followed by hybridization with p53 polymorphic probe showed the presence of a 12 kb fragment in majority of the tumor tissues. However, some of the tissues showed an additional 9 kb fragment along with the 12 kb fragment. Both the fragments represent allelic variants of the p53 gene and the additional Bgl II enzyme site was mapped in the intron 1 (48). The wild type p53 gene expression and function was not affected due to the presence of this additional Bgl II site and the p53 protein also had no differences except for the substitution of arginine for proline at the 72th position in the proline rich region (45,48) (Figure 3,4,5).
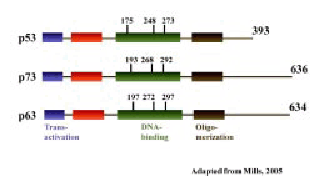
Figure 4 : The p53 family, Diagramatic repersentation of the structural homology between p53, p63 and p73 proteins. The numbers above represent the amino acids
Structure of the p53 Protein
The p53 gene encodes for a protein of 393 amino acids with a molecular weight of 53 kDa (49). Based on its structure and function the p53 protein has been divided into three distinct domains, (a) the transcriptional activation domain at the amino terminal or N-terminus, (b) the central sequence-specific DNA binding domain and (c) the multifunctional basic carboxy terminal or C-terminal domain (Figure 3)
Isoforms of p53
The human p53 gene contains alternative promoters and transcribes multiple splice variants or isoforms and these are expressed in normal human tissue in a tissue-dependent manner and they are also found in some tumor tissues (21,50). Instead of the normal pattern of splicing between exons 9 and 10 that occurs to generate full-length p53, the β and γ isoforms are devoid of amino acids encoded by exon 10, and instead are equipped with 10 and 15 novel amino acids, respectively, due to cryptic splice sites located within intron 9 that are promptly followed by premature stop codons. Thus, the β isoform terminates with the amino acids ‘DQTSFQKENC and the γ isoform ends with the amino acids ‘MLLDLRWCYFLINSS’ (50). These isoforms are speculated to have altered functions (21,50).
The p53 Family
Two members of the p53 family, p63 and p73 have been identified. The structures of the p63 and p73 genes are more similar to one another than to p53. Similar to p53, both p63 and p73 can form homo-oligomers, bind DNA, activate transcription from p53-responsive genes, and induce apoptosis (51–53) (Figure 4).
Posttranslational Modifications, Stabilization and Degradation of p53 Protein
Phosphorylation of p53 protein has been shown to increase its sequence-specific DNA binding (54). Approximately 17 phosphorylation sites have been identified in human cells following DNA damage induced by ionizing radiation or ultraviolet (UV) -light radiation. In humans these residues include serines 6,9,15, 20, 33, 37, 46 and threonines 18, 81 in the N-terminal region; Ser 315 and Ser 392 in the C-terminal domain; and Thr 150, Thr 155, Ser 149 in the central core (55,56). In addition, Thr 55, Ser 376 and Ser 378 are reported to be constitutively phosphorylated in unstressed cells. Phosphorylation at Ser 33, 46 and Thr 81 has been shown to increase the half-life of the p53 protein and hence stabilize the same. Acetylation of p53 is suggested to be important for p53 stability and transcriptional activation (55,56). Under normal conditions the p53 protein is a latent short-lived protein with a half-life of 6 to 20 minutes (57). Having a short half-life the p53 protein is normally maintained at low levels in unstressed cells by continuous ubiquitylation and subsequent degradation via MDM2 and the 26S proteasome (45,55,57) (Figure 5).
Cellular Functions of p53
The p53 pathway is composed of hundreds of genes and their products that respond to a wide variety of intrinsic and extrinsic stress signals such as DNA damage, oncogene activation, hypoxia, cellular ribonucleotide depletion, mitotic spindle damage and nitric oxide (NO) production (58). These stress signals all impact upon the cellular homeostatic mechanisms that monitor and control the fidelity of DNA replication, chromosome segregation and cell division (59). Among the stresses that activate the p53 protein is damage to the integrity of DNA in a cell (45,57) (Figure 6).
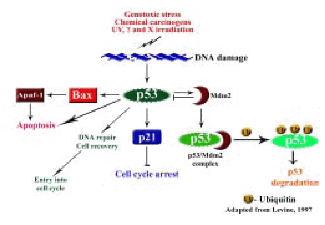
Figure 6: The p53 pathway
Role of p53 in Cell Cycle Arrest
p53 functions as a cell cycle checkpoint protein and plays an important role in the induction of cell cycle arrest in response to DNA damage (Figure 5). p53 has been implicated in both the G1-S phase and the G2-M phase checkpoints of the cell cycle (60,61). The Rb protein regulates the restriction point or start-signal for cell cycle progression. In response to DNA damage p53 is activated which in turn transactivates one of its downstream genes p21 (WAF1, Cip-1) (62). p21 binds to a number of cyclin and CDK complexes and inhibits the kinase activity thus preventing the phosphorylation of Rb resulting in cell cycle arrest. Thus, through the inhibition of CDKs, p21 acting downstream of p53 arrests the cells at the G1-S transition (45,63) (Figure 6).
p53 and Apoptosis
p53 is known to promote apoptosis through transcription-dependent and independent mechanisms that act in concert to ensure that the cell death program proceeds efficiently (45,64). Two major pathways trigger the apoptosis program: the death receptor induced extrinsic pathway and the mitochondria apoptosome mediated intrinsic pathway. The link between p53-mediated transactivation and apoptosis comes from its ability to control the transcription of the pro-apoptotic members of the Bcl-2 family, Bax, as well as the BH3-only members Puma, Noxa, and Bid. This increases the ratio of pro-apoptotic to anti-apoptotic Bcl-2 proteins, thereby favoring the release of cytochrome c from mitochondria into the cytoplasm leading to caspase activation and apoptosis. Many studies have indicated that p53-mediated apoptosis proceeds primarily through the intrinsic pathway. The extrinsic pathway is also regulated by p53 but the overall contribution of this regulation to p53-mediated cell death is poorly understood (45,64) (Figure 6).
Transcription-independent activities of p53 in apoptosis
Transcription independent pro-apoptotic functions of p53 have been proposed many years ago and recently have been shown to facilitate cell death by genotoxic agents. It has been suggested that the transcription-independent pro-apoptotic activities of p53 is due to its ability to modulate the functions of the proteins involved in the apoptotic machinery (45,65).
Antiapoptotic effects of p53
p53 has been observed to possess antiapoptotic capabilities under a variety of conditions. The cells lacking p53 are sometimes more sensitive to apoptosis than their p53-proficient counterparts. Studies have implicated the wild type p53 protein in protection against cell death. It has been suggested that this mechanism involves the ability of p53 to bring about more effective DNA repair. The ability of p53 to turn on several antiapoptotic genes in addition to many proapoptotic targets suggests a possibility that the decision of life or death of a cell is determined by p53 (45,65).
p53 in Senescence and Aging
p53 is a critical regulator of the senescence response to a variety of signals including short telomeres, DNA damage, oncogenes and overexpressed tumor suppressor genes. Early indications of the importance of p53 in cellular senescence came from studies using the SV-40 large-T antigen, which binds and inactivates p53. The T-antigen extended the replicative life span of cultured human fibroblasts and also stimulated postmitotic senescent cells to initiate DNA replication. Human cells that over express oncogenic ras or E2F1 or the pl4ARF or PML tumor suppressors fail to undergo a senescence-arrest if p53 function is defective. These studies clearly indicate a role for wild type p53 in senescence (66).
Another important function with which p53 has been associated in the recent times is organismal aging (67). Using mice model association between increased wild type p53 activity and premature organismal aging was demonstrated. In contrast to these studies it has also been illustrated that under specific circumstances excess levels of wild type p53 can protect mice against cancer and aging. A hint that reduced level of p53 could potentially increase longevity has also been observed in p53 heterozygous mice that could evade tumor formation. These mice were observed to live longer as compared to their wild type counterparts (67).
p53 in the Maintenance of Genetic Stability
Consistent with the role for p53 in protecting genomic integrity, fibroblasts from p53 deficient mice have demonstrated chromosomal abnormalities that appear at early passage in homozygous null fibroblast and at later passage in heterozygous fibroblasts. Aneuploidy and chromosome instability have also been demonstrated in p53 null mice. The cellular DNA replication is shown to be error-prone and the DNA repair genes correct these errors. When these DNA repair genes are inactivated the cells accumulate errors in genes leading to genetic instability and predispose the cells to tumorigenesis. Two specific genes involved in global genomic repair DDB2 and XPC are shown to contain p53 binding consensus sequences. Products of these genes p48 and XPC respectively have been shown to increase in the presence of wild type p53 in response to UV irradiation. Under these conditions the global genomic repair of these cells were shown to be enhanced. Deficiency in base excision repair mechanism has also been observed in cells lacking wild type p53 indicating a possible role for p53 in this mechanism of DNA repair (45,59) (Figure 6).
p53 in Differentiation and Embryonic Development
p53 deficient mice are reported to exhibit developmental abnormalities of the nervous system at a high frequency. p53 deficient mice embryos have been shown to be defective in neural tube closure resulting in an overgrowth of neural tissue in the region of the mid-brain resulting in a condition known as exencephaly. High-frequency of developmental abnormalities including neural tube defects, ocular abnormalities and defects in upper incisor tooth formation have been reported in p53-deficient mice and these abnormalities were found predominantly in females. Studies have suggested the presence of a p53-dependent “guardian” in the embryonic or fetal tissues which plays an “embiyo-protective” role by aborting cells bearing teratogenic DNA damage and hence prevent off-springs with developmental abnormalities. p53 is also suggested to play a key role in protecting embryos against diverse environmental stresses (68).
Recent studies have also implicated p53 in adult neurogenesis. Neuronal-turnover is a two-step process wherein an excess of neuronal progenitors are generated, only few of the progenitors differentiate into fully functional neurons while the excessive progenitor cells are eliminated. Alteration in the p53 activity has been shown to upset this fine balance by affecting the rate of cell proliferation but not the rate of cell death in the neurogenic regions of adult brain. Genetically engineered mice with increased p53 activity have shown premature loss of neurogenic capacity. A link between the premature loss of neurogenic capacity and accelerated organismal aging has been demonstrated (69).
Role of p53 in Carcinogenesis
Identification of germ-line p53 mutations in the familial Li-Fraumeni syndrome associated with an early onset of various cancers strongly suggested a role for the p53 gene in tumorigenesis. The fact that p53 null mice were highly prone to tumors further emphasized that the loss of p53 function plays an important role in tumorigenesis. p53 is the most commonly mutated gene in human malignancies, with mutations reported in more than 50% of all cancers (40,45) (Figure 6). In the latest released version of the IARC p53 mutation database there are 19,809 somatic mutations of p53 reported.
The wild type p53 protein functions optimally when it binds to DNA as a wild type p53 tetramer. One mutant p53 protein can disturb a functional tetramer and is therefore able to override the function of the wild type p53 protein. This is known as the ‘dominant negative’ means of inactivation (71). In this mechanism mutation in only one allele of the p53 gene is sufficient for the functional inactivation of both the alleles, although loss of the second, wild type allele may further contribute to oncogenesis (72).
p53 loses its tumor suppressor function as a consequence of mutation as most p53 mutants have impaired function with respect to sequence-specific transactivation of genes. Certain types of p53 mutations exert functions that the wild type p53 does not and hence are called as the “gain-of-function” mutants (73). Mutant p53 alleles expressed in cell lines lackingp53 resulted in enhanced tumorigenic potential, metastatic capacities and a shorter survival in mice (74). Gain-of-function mutants of p53 are known to exert oncogenic effects through the induction of increased expression of a diverse group of genes either directly or indirectly (73).
Mutant p53 proteins are shown to be intensely phosphorylated and acetylated at sites that are well known to be involved in the stabilisation of wild type p53. Such altered p53 proteins have been shown to facilitate accumulation of dysfunctional, mutant p53 in the nucleus, where it can exert oncogenic functions (56). Intense acetylation of the mutant p53 proteins at the lysine residues 320, 373, 382 and significant phosphorylation at Ser 392, Ser 15, Thr 81 has been reported in tumor-derived cell lines (75). Phosphorylation at these residues is suggested to stabilise the mutant p53 protein by inhibiting its degradation and facilitating its accumulation in the nucleus. Greater level of phosphorylation at Ser 6, 15, 37 and Thr 81 of wild type p53 in human tumors has also been reported but the significance of these observations is not yet clear (75). Studies on the UV-light-induced mouse tumors have shown that mutant p53 protein in these tumors is constitutively phosphorylated at Ser 15, localized in the nucleus and is resistant to MDM2-mediated degradation (76). These data indicate that phosphorylation of mutant p53 at Ser 15 contributes to its increased stability and to its oncogenic activities (Bode and Dong, 2004). Phosphorylation of p53 at Ser 392 has been reported to stabilize the tetramer formation (77). In human transitional cell carcinomas 60% of the tumors with accumulated mutant p53, but none with wild type p53,showed phosphorylation of Ser 392 (78). Loss of phosphorylation at the Ser 392 is reported to increase the tumorigenic potential of mutant p53 and also inhibit p53-induced apoptosis suggesting its importance for the oncogenic function of mutant p53 (79). Ser 392 phosphorylation has been suggested to be an early event in the pathogenesis of squamous cell carcinoma (80). Higher levels of Ser 392 phosphorylated p53 have been associated with higher proliferation, tumor progression and poor prognosis of human esophageal squamous cell carcinoma (80). Increased Ser 392 phosphorylation of mutant p53 has been frequently reported in transitional cell carcinoma (TCC). It has been suggested to promote the dominant negative effects of the mutant p53 through hetero-oligomerization, thereby contributing to the proliferation and aggressive behavior of these tumors (78). Increased percentage of Ser 392 phosphorylated form of p53 protein was found in human high grade gliomas as compared to the lower grade astrocytomas (21). In addition, Ser 392 phosphorylation of the p53 protein was eported to occur in human vestibular schwannomas (VS) in an age dependentmanner(16,17).
Alterations of the p53 Gene in Human Brain Tumors
Alteration of the p53 gene has been reported in all grades of gliomas and studies have suggested that p53 plays a role both in the formation of low-grade disease and in the progression towards tumors of higher histological grade (9–11,21). Mutations in the p53 gene have been reported in more than 50% of gliomas (81). The fact that patients with Li-Fraumeni syndrome, who inherit germline mutations in the p53 gene are predisposed to the development of brain tumors early in life and that gliomas are part of the tumor spectrum suggested a causal role for p53 inactivation in gliomagenesis (82). Addition of wild type p53 to the glioblastoma multiforme cell lines lead to reversal of their malignant phenotype confirming a role for p53 in the astrocytic tumor development (83). p53 mutations in astrocytomas were first described in late eighties and were followed by a more extensive analysis of the gene mutations and p53 protein alterations in adult astrocytomas. Accumulation of wild type p53 protein has been reported in astrocytomoas (84). The loss of p53 gene and LOH at this locus have also been implicated in the progression of gliomas (21,85). In the anaplastic astrocytomas grade III more than 60% cases have been reported to harbor p53 gene mutations (86). p53 has been suggested to play a role in the progression of secondary glioblastomas. The p53 protein is detected in approximately 15 to 40% of low grade astrocytomas, 35 to 60% of anaplastic astrocytomas, and 45 to 70% of glioblastomas (87). Malignant progression of astrocytic neoplasms has been associated with increasing expression of p53 protein (21,87).
p53 as a Therapeutic Target
Mutations at p53 gene locus are present in every second human tumor and during malignant transformation p53 or p53-pathway related molecules are known to be frequently disabled. Therefore, inactivation of p53 is critical for tumorigenesis. Since the normal function of p53 is critical to the regulation of cell cycle arrest and apoptosis, restoration of the p53 pathway has been the logical strategy for the treatment of cancer (88). Molecular therapeutic strategies to normalize p53 signaling in cells with mutant p53 include pharmacologic rescue of mutant protein, gene therapy, small-molecule agonists of downstream inhibitory genes, antisense and use of oncolytic viruses. Other strategies include activation of normal p53 pathway, inhibition of mdm2-mediated degradation of p53 and blockade of p53 nuclear export (89). p53 is one of the important molecular targets in radiation oncology. It is an important molecule that determines tissue-specific radiosensitivity. As a consequence the p53 pathway can be exploited to enhance cancer therapies especially when the tumors are caused by DNA damaging agents (90). Ad-p53 has been suggested to be most effective against gliomas when combined with radiation therapy and chemotherapy. Despite these encouraging findings, use of p53 gene therapy in patients has its constraints in the efficiency of gene delivery and therefore development of novel methods ensuring the gene distribution throughout the tumor is required. A recent study on the child hood tumor retinoblastoma has revealed deregulation of the p53 pathway due to over expression of MDMX, a negative regulator of p53. MDMX has therefore been identified as a specific chemotherapeutic target for treatment of these tumors (91).
Current Status
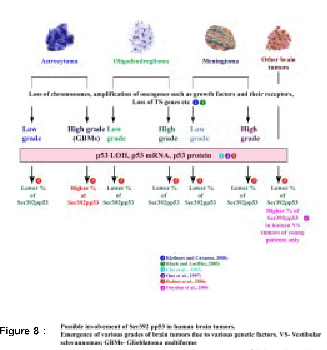
Human brain tumors arise as a consequence of accumulation of multiple genetic alterations in brain cells, these cells become tumorigenic and further progression of these tumors lead to malignant phenotype. Brain tumors are heterogeneous because of the various cell types from which they initiate and are classified accordingly (2,92) (Figure 7). Wide range of genetic alterations is acquired during human brain tumor development and also during their further progression to higher histological grades. The various genetic alterations that characterize low-grade and high-grade tumors are as shown in Figure 8 (2,10,16,17,21,87,93).
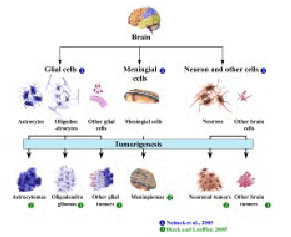
Figure 7 : Various types of human brain tumors and their posible orgins.
The wild type p53 protein is known to function as a cell cycle checkpoint protein at G1 to S and G2 to M transition phases of the cell cycle. Under normal conditions p53 gene is known to be expressed at lower levels in all tissues. In response to various cellular stresses that could cause DNA damage the level of p53 protein is reported to increase in these cells. As a response to DNA damage, p53 is known to induce cell cycle arrest which could allow repair of the damaged DNA. If the damage is irreparable then the p53 protein is known to induce apoptosis and thereby eliminate cells that contain damaged DNA (45). The increased p53 protein during such a process is then rapidly degraded by binding to MDM2, a primary negative regulator of p53 protein, which maintains a controlled p53 protein level in normal cells (45,94) (Figure 9a). Mutations at the p53 gene locus result in an altered or mutant p53 protein, which is resistant to MDM2-mediated degradation and hence results in the accumulation of the same in the cells carrying these mutations (45,95,96). Wild type p53 is a tumor suppressor and it is also a wen-known DNA binding protein, which is known to bind to DNA as a tetramer. Presence of one mutant p53 allele and one wild type allele results in a hetero-tetramer complex containing both mutant and wild type p53 protein. The mutant p53 protein in such a complex is known to inactivate the wild type p53 protein in a ‘dominant negative’ fashion resulting in the loss of wild type p53 functions including the tumor suppressor function, cell cycle arrest and apoptosis. Presence of such a p53 tetramer in the cell with altered function could eventually lead to tumor development (45,95,96) (Figure 9b). If both the alleles of the p53 gene are altered it would result in a mutant tetramer which could lead to tumorigenesis (96) (Figure 9c).
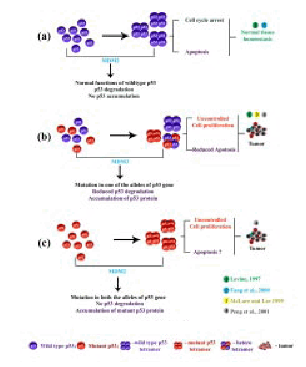
Figure 9: Functions of wild type and implant p53 protein
LOH at the p53 gene locus, increased level of p53 mRNA and protein in all the human brain tumor types and tumors of all grades have been reported (21). The increased levels of p53 mRNA and protein observed in all the human brain tumor types could be due to deregulation of p53 pathways (97). Therefore, results from these studies are suggestive of a role for the p53 gene in the overall development of human brain tumors (21) (Figure 8,9).
The observed increase in the levels of p53 protein in all the human brain tumor samples analysed could be due to a defective MDM2-mediated p53 degradation pathway in these tumors (98). Recent studies have reported increased apoptosis in neuronal cells with increased levels of wild type p53 protein (97). Therefore, it is conceivable that the increased level of p53 protein could lead to increased apoptosis in slow growing tumors such as meningiomas. Meningiomas are reported to be mostly benign and slow growing tumors (81). The slow growing behavior of meningiomas could possibly be due to a higher rate of apoptosis induced by the increased p53 protein levels, which in turn could be a limiting factor in determininone growth rate of these tumors (Figure 10).
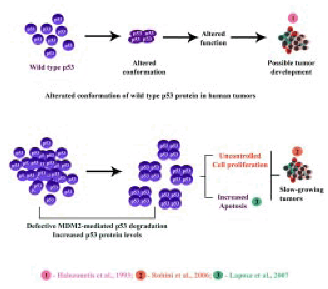
Figure 10 : Increased level of p53 protein in slow growing tumors such as menisgiomax.
Increased percentage of Ser 392 phosphorylated form of p53 protein was found only in the high-grade gliomas of grade III and IV (GBMs). To the contrary, lower percentage of Ser 392 phosphorylated form of p53 was present in all the low-grade gliomas and meningiomas of all histological grades as well as in the other brain tumor types (21) (Figure 11). Presence of increased percentage of Ser 392 phosphorylated form of p53 protein has been reported in human vestibular schwannomas (VS) of young patients only (16,17). The VS tumors in the young patients have been reported to be highly proliferative compared to that of the older patients (99). Therefore, the observed increase in the percentage of Ser 392 phosphorylated form of p53 protein only in the high-grade gliomas could be indicative of high proliferative potential of these tumors (21). Phosphorylation at the Ser 392 residue of the p53 protein has been reported to stabilize the p53 tetramers (77). It appears that the increased percentage of Ser 392 phosphorylated form of p53 protein could lead to the formation of highly stable tetramers, which could be resistant to MDM2-mediated degradation (Figure 11). This could result in accumulation of higher levels of Ser 392 phosphorylated form of p53 protein in these high-grade gliomas (Figure 11). The wild type p53 protein has been reported to adopt a mutant-like conformation when it binds to DNA (100) (Figure 10). It is conceivable that the presence of higher percentage of Ser 392 phosphorylated form of p53 protein in the high-grade gliomas could have acquired an altered conformation. The function of wild type p53 is suggested to be regulated via its ability to adopt distinct conformation (100) and therefore an altered conformation could result in an altered function, which in turn could aid in tumor development (Figure 11). Increased Ser 392 phosphorylation of mutant p53 protein has been suggested to promote its dominant negative effects through hetero-oligomerization, thereby contributing to the proliferation and the aggressive behavior of transitional cell carcinomas (TCC) (78). Wild type p53 protein is also reported to undergo phosphorylation at the Ser 392 residue (55). The high-grade gliomas, particularly the GBMs are known to be biologically one of the most aggressive tumors with a survival period of less than 1 year for the patients from the time of diagnosis (82). Therefore, presence of higher percentage of Ser 392 phosphorylated form of p53 protein only in the high-grade gliomas could be associated with the aggressive behavior of these tumors (21) (Figure 11). Emphasizing this conclusion further is the presence of lower percentage of Ser 392 phosphorylated form of p53 protein in all the low-grade gliomas as well as meningiomas of all grades and the other human brain tumor types. Meningiomas are reported to be mostly benign, slow growing and biologically less-aggressive tumors (2) (Figure 10,11). Presence of higher percentage of Ser 392 phosphorylated form of p53 protein in the high-grade tumors could be suggestive of highly aggressive and fast growing tumors with high proliferative potential, decreased apoptosis and enhanced metastatic potential (21) (Figure 10,11). Moreover, presence of lower percentage of Ser 392 phosphorylated form of p53 in slow growing tumors like meningiomas is suggestive of its role in less-aggressive tumors with low proliferative potential. It is conceivable that these slow growing tumors could have increased rate of apoptosis, probably p53-mediated apoptosis. Thus, presence of increased level of p53 protein could have a role in p53-mediated apoptotic pathway in these slow-growing tumors (21) (Figure 11).
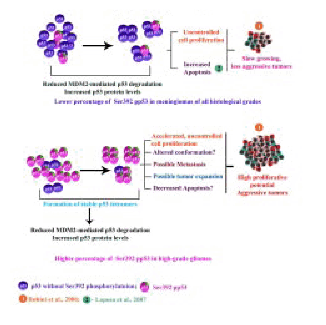
Figure 11 : Possible role of Ser 392 Phosphorylation p53 in the growth of human brain tumors.
Conclusion
Human brain tumors are one of the most difficult to manage and treat. Research in the past four decades resulted in no improvement in the survival of these patients. Therefore, development of novel approaches is essential to manage and treat these tumors. The p53 gene is one of the most important human tumor suppressor gene which affects both cell growth and cell death. Experimental evidence suggests that the function of p53 could be modulated at various levels – gene structure, expression, level of mRNA and protein, protein conformation and post-translational modification. Apart from point mutations, alteration at any of these levels could also affect the function of the protein and hence could lead to tumor development. In addition, presence of p53 isoforms along with the wild type p53 could also affect the tumor suppressor functions of the latter and could lead to tumor progression. Thus, in addition to other genetic parameters it is important to evaluate the p53 gene structure and expression and the protein status, posttranslational modification in particular, and presence of isoforms, if any, in individual tumor as this information could help in the development of custom-made protocols to manage and treat various human brain tumor types.
Acknowledgement
We thank all the faculty members of the NIMHANS Neurosurgery department for providing the tumor tissues which were used in some of the publications sited in this review. Some of the results presented in this manuscript are from the research project funded by the Department of Biotechnology (DBT), Government of India, to Dr. R. Gope (Principal Investigator), project number BT/PRO 703/MED/09/133/97. Financial assistance in the form of Junior and Senior Research Fellowships (JRF, SRF) to RK and MJ from UGC are gratefully acknowledged.
References
1. Ellison D, Love S, Chimelli L, Harding BN, Lowe S, Vinters HV (eds). Neuropathology: A reference text of CNS pathology 2nd edition, London,Mosby © Elsevier Limited, 2004.
2. Black PM, Loeffler J (eds). Cancer of the Nervous System, 2nd ed. Philadelphia, Lippincort, Williams and Wilkins, 2005.
3. Collins VR Brain Tumors: Classification and Genes. J Neurol Neurosurg Psychiatry 2004; 75:2–11.
4. Kaye A, Laws E Jr. Brain Tumors: An Encyclopedic Approach. 2nd ed. New York, NY, Churchill Livingstone; 2001.
5. Ohgaki H, Kleihues R Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 2005; 64: 479–89.
6. Huang K, Whelan EA, Ruder AM, et al. The Brain Cancer Collaborative Study Group. Reproductive factors and risk of glioma in women. Cancer Epidemiol Biomarkers Prev 2004; 13: 1583–88.
7. DeAngelis LM, Gutin PH, Leibel SA et al. Intracranial tumors diagnosis and treatment. London, UK, Martin Dunitz, 2002.
8. Juven Y, Sadetzki S. A possible association between ionizing radiation and pituitary adenoma: a descriptive study. Cancer 2002; 95: 397–403.
9. Kleihues P Burger PC, Scheithauer BW. Histological typing of tumors of the central nervous system. Berlin: Springer-Verlag, 1993.
10. Kleihues P, Cavenee WK (eds) World Health Organisation: Classification of tumors, Lyon, France, IARC Press, 2000.
11. Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol 2002; 61: 215–29.
12. Westermark B, Nister M. Molecular genetics of human glioma. Curr Opin Oncol. 1995; 7: 220–25.
13. Gutmann DH, Donahoe J, Brown T, et al. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol 2000; 26: 361–67.
14. Hill JR, Kuriyama N, Kuriyama H, et al. Molecular genetics of brain tumors. Arch Neurol 1999; 56: 439–41.
15. Rasheed BK, McLendon RE, Herndon JE, et al. Alterations of the TP53 gene in human gliomas. Cancer Res 1994; 54: 1324–30.
16. Dayalan AHPP, Mathivanan J, Rohini K, et al. Age dependent phosphorylation and deregulation of p53 human vestibular schwannomas. Mol Carcinogenesis 2006; 45: 38–46.
17. Dayalan AHPP, Rohini K, Mathivanan J, et al. The p53 gene and human vestibular schwannomas. Ann Neurosa 2006; 13: 77–91.
18. Dayalan AHPP, Thomas R, Mathivanan J, et al. The tumor suppressor gene retinoblastoma (RB1) in human vestibular schwannomas. Ann Neurosci 2006; 13: 113–24.
19. Mathivanan J, Rohini K, Gope ML, et al. Altered structure and deregulated expression of the tumor suppressor gene retinoblastoma (RB1) in human brain tumors. Mol Cell Biochem 2007; 302: 67–77.
20. Mathivanan J, Rohini K, Gope ML, et al. Possible role of the tumor suppressor gene retinoblastoma (RB1) in human brain tumor development. Ann Neurosci 2007; 14: 72–82.
21. Rohini K, Mathivanan J, Antony Herold Prabhu PD, et al. Loss of heterozygosity of the p53 gene and deregulated expression of its mRNA and protein in human brain tumors. Mol Cell Biochem 2007; 300: 101–11.
22. Thomas R, Antony Herold Prabhu PD, Mathivanan J, et al. Altered structure and expression of RB1 gene and increased phosphorylation of pRb in human vestibular schwannomas. Mol Cell Biochem 2005; 271: 113–21.
23. Hermanson M, Funa K, Hartman M, et al. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res 1992; 52: 3213–19.
24. Watanabe K, Tachibana O, Sata K, et al. Over expression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol 1996; 6: 217–23.
25. James CD, He J, Carlbom E, et al. Chromosome 9 deletion mapping reveals interferon alpha and interferon beta-1 gene deletions in human glial tumors. Cancer Res 1991; 51: 1684–88.
26. Ueki K, Ono Y, Henson JW et al. CDKN2/ pl6 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res 1996; 56: 150–53.
27. Henson JW, Schnitker BL, Correa KM, et al. The retinoblastoma gene is involved in malignant progression of astrocytomas. Ann Neurol 1994; 36: 714–21.
28. Mantani M, Israel MA: Brain Tumors, In: Schwab (ed). Encyclopedic reference of cancer. Springer Verlag, Italy, 2001; 131–34.
29. Preul MC, Caramanos Z, Collins DL, et al. Accurate, noninvasive diagnosis of human brain tumors by using proton magnetic resonance spectroscopy. Nat Med 1996; 2: 323–25.
30. Ostergaard L, Hochberg FH, Rabinov JD, et al. Early changes measured by magnetic resonance imaging in cerebral blood flow, blood volume, and blood-brain barrier permeability following dexamethasone treatment in patients with brain tumors. J Neurosurg 1999; 90: 300–305.
31. Flickinger JC. Radiotherapy and radiosurgical management of brain metastases. Curr Oncol Rep 2001; 3: 484–89.
32. Engelhard HH. Gene therapy for brain tumors: the fundamentals. Surg Neurol 2000; 54: 3–9.
33. Moreira AL, Friedlander DR, Shif B, et al. Thalidomide and a thalidomide analogue inhibit endothelial cell proliferation in vitro. J Neurooncol 1999;43: 109–14.
34. Yih TC, Al-Fandi M. Engineered nanoparticles as precise drug delivery systems. J Cell Biochem 2006; 97: 1184–90.
35. Kano MR, Bae Y, Iwata C, et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF- signaling. Proc Natl Acad Sci USA 2007; 104: 3460–65.
36. Reddy GR, Bhojani MS, McConville R et al. Vascular targeted nanoparticles for imaging and treatment of brain tumors. Clin Cancer Res 2006; 12: 6677–86.
37. Mowat M, Cheng A, Kimura N, et al. Rearrangements of the cellular p53 gene in erythroleukaemic cells transformed by Friend virus. Nature 1985; 314: 633–36.
38. Eliyahu D, Goldfinger N, Pinhasi-Kimhi O, et al. Meth A fibrosarcoma cells express two transforming mutant p53 species. Oncogene 1988; 3: 313–21.
39. Finlay CA, Hinds PW, Tan TH, et al. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol Cell Biol 1988; 8: 531–39.
40. Hollstein M, Sidransky D, Vogelstein B, et al. p53 mutations in human cancers. Science 1991; 253: 49–53.
41. Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991; 351: 453–56.
42. McBride OW, Merry D, Givol D. The gene for human p53 cellular tumor antigen is located on chromosome 17 short arm (17pl3). Proc Natl Acad Sci USA 1986; 83: 130–34.
43. Benchimol S, Lamb P, Crawford LV et al. Transformation associated p53 protein is encoded by a gene on human chromosome 17. Somat Cell Mol Genet 1985; 11: 505–10.
44. Lamb P, Crawford L. Characterization of the human p53 gene. Mol Cell Biol. 1986; 6: 1379–85.
45. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–31.
46. Matlashewski G, Lamb P, Pim D, et al. Isolation and characterization of a human p53 cDNA clone: expression of the human p53 gene. EMBO J. 1984; 3: 3257–62.
47. Rogel A, Popliker M, Webb CG, et al. p53 cellular tumor antigen: analysis of mRNA levels in normal adult tissues, embryos, and tumors. Mol Cell Biol 1985; 5: 2851–55.
48. Buchman VL, Chumakov PM, Ninkina NN et al. A variation in the structure of the protein-coding region of the human p53 gene. Gene 1988; 70: 245–52.
49. Vogelstein B, Kinzler KW p53 function and dysfunction. Cell 1992; 70: 523–26.
50. Bourdon JC, Fernandes K, Murray-Zmijewski F et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev 2005; 19: 2122–37.
51. Jost CA, Marin MC, Kaelin WG Jr. p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature 1997; 389: 191–94.
52. Kaelin WG Jr. The emerging p53 gene family. J Natl Cancer Inst 1999; 91: 594–98.
53. Mills AA. P53: Link to the past, bridge to the future. Genes Dev 2005; 19: 2091–99.
54. Hupp TR, Lane DP Regulation of the cryptic sequence-specific DNA-binding function of p53 by protein kinases. Cold Spring Harb Symp Quant Biol 1994; 59: 195–206.
55. Appella E, Anderson CW Signalling to p53: breaking the posttranslational modification code. Pathol Biol Paris 2000; 48: 227–45.
56. Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 2004; 4: 793–805.
57. Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ 2006; 13: 1027–36.
58. Jin S, Levine AJ. The p53 functional circuit. J Cell Sci 2001; 114: 4139–40.
59. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408: 307–10.
60. Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell 1990; 62: 671–80.
61. Stewart N, Hicks GG, Paraskevas F, et al. Evidence for a second cell cycle block at G2/M by p53. Oncogene 1995; 10: 109–15.
62. El-Deiry WS, TokinoT, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–25.
63. Slebos RJ, Lee MH, Plunkett BS, etal. p53-dependent G1 arrest involves pRB-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. .Proc Natl Acad Sci USA 1994; 91: 5320–24.
64. Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene 2003; 22: 9030–40.
65. Oren M. Decision making by p53: life, death and cancer. Cell Death Differ 2003; 10: 431–42.
66. Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. Eur J Biochem 2001; 268: 2784–91.
67. Donehower LA. Does p53 affect organismal aging? J Cell Physiol 2002; 192: 23–33,
68. Hall PA, Lane DP Tumor suppressors: a developing role for p53? Curr Biol 1997; 7: 144–47.
69. Medrano S, Scrable H. Maintaining appearances--the role of p53 in adult neurogenesis. Biochem Biophys Res Commun 2005; 331: 828–33.
70. Olivier M, Hussain SP, Caron de Fromentel C, et al. TP53 mutation spectra and load: a tool for generating hypotheses on the etiology of cancer. IARC Sci Publ 2004; 157: 247–70.
71. Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell. 1991; 65: 765–74.
72. Weinberg RA. Oncogenes and tumor suppressor genes. CA Cancer J Clin 1994; 44: 160–70.
73. van Oijen MG, Slootweg PJ. Gain-of-function mutations in the tumor suppressor gene p53. Clin Cancer Res 2000; 6: 2138–2145.
74. Dittimer D, Pati S, Zambatti G, et al. Gain of function mutations in p53. Nature Genet 1993; 4: 42–6.
75. Minamoto T, Buschmann T, Habelhah H, et al. Distinct pattern of p53 phosphorylation in human tumors. Oncogene 2001; 20: 3341–47.
76. Melnikova VO, Santamaria AB, Bolshakov SV, et al. Mutant p53 is constitutively phosphorylated at Serine 15 in UV-induced mouse skin tumors: involvement of ERK1/2 MAP kinase. Oncogene 2003; 22: 5958–66.
77. Sakaguchi K, Sakamoto H, Lewis MS, et al. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 1997; 36: 10117–24.
78. Furihata M, Kurabayashi A, Matsumoto M, et al. Frequent phosphorylation at serine 392 in over expressed p53 protein due to missense mutation in carcinoma of the urinary tract. J Pathol 2002; 197: 82–8.
79. Yap DB, Hsieh JK, Zhong S, et al. Ser392 phosphorylation regulates the oncogenic function of mutant p53. Cancer Res 2004; 64: 4749–54.
80. Matsumoto M, Furihata M, Kurabayashi A, et al. Prognostic significance of serine 392 phosphorylation in over expressed p53 protein in human esophageal squamous cell carcinoma. Oncology 2004; 67: 143–50.
81. Nagane M, Huang HJ, Cavenee WK. Advances in the molecular genetics of gliomas. Curr Opin Oncol 1997; 9: 215–22.
82. Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev 2001; 15: 1311–33.
83. Van Meir EG, Roemer K, Diserens AC, et al. Single cell monitoring of growth arrest and morphological changes induced by transfer of wild-type p53 alleles to glioblastoma cells. Proc Natl Acad Sci USA 1995; 92: 1008–12.
84. Rubio MP von Deimling A, Yandell DW, et al. Accumulation of wild type p53 protein in human astrocytomas. Cancer Res 1993; 53: 3465–67.
85. Fueyo J, Gomez-Manzano C, Yung WK, et al. Targeting in gene therapy for gliomas. Arch Neurol 1999; 56: 445–48.
86. Ichimura K, Bolin MB, Goike HM, et al. Deregulation of the pl4ARF/ MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res 2000; 60: 417–24.
87. Cho MY, Jung SH, Kim TS. P53 protein over expression in astrocytic neoplasms. Yonsei Med J 1995; 36: 521–26.
88. Stoklosa T, Golab J. Prospects for p53-based cancer therapy. Acta Biochim Pol 2005; 52: 321–28.
89. Lane DP, Hupp TR. Drug discovery and p53. Drug Discov Today 2003; 8: 347–55.
90. Schmidt-Ullrich RK. Molecular targets in radiation oncology. Oncogene 2003; 22: 5730–33.
91. Laurie NA, Donovan SL, Shih CS, et al. Inactivation of the p53 pathway in retinoblastoma. Nature 2006; 444: 61–6.
92. Noback CR, Strominger NL, Demarest RJ, Ruggiero DA (eds). The human nervous system. Totowa, NJ, Humana Press, 2005.
93. Ono Y, Tamiya T, Ichikawa T, et al. Accumulation of wild-type p53 in astrocytomas is associated with increased p21 expression. Acta Neuropathol Berl 1997; 94: 21–7.
94. Fang S, Jensen JP, Ludwig RL, et al. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 2000; 275: 8945–51.
95. McLure KG, Lee PW. P53 DNA binding can be modulated by factors that alter the conformational equilibrium. EMBO J 1999; 18: 763–70.
96. Peng Y, Chen L, Li C, et al. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J Biol Chem 2001; 276: 40583–90.
97. Laposa RR, Huang EJ, Cleaver JE. Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc Natl Acad Sci USA 2007; 104: 1389–94.
98. Yamauchi M, Suzuki K, Kodama S, et al. Abnormal stability of wild-type p53 protein in a human lung carcinoma cell line. Biochem Biophys Res Commun 2005; 330: 483–88.
99. Baser ME, Makariou EV, Parry DM. Predictors of vestibular schwannoma growth in patients with neurofibromatosis Type 2. J Neurosurg 2002; 96: 217–22.
100. Halazonetis TD, Davis LJ, Kandil AN. Wild-type p53 adopts a ‘mutant’-like conformation when bound to DNA. EMBO J 1993; 12: 1021–28.
(c) Annals of Neurosciences.All Rights Reserved